Une nouvelle piste dans la compréhension du syndrome de l'X fragile
16/09/2020
Izabela Sumara et son équipe « Cycle cellulaire et signalisation de l'ubiquitine » de l’Institut de génétique et de biologie moléculaire et cellulaire (IGBMC) ont découvert que la famille des protéines liées au syndrome de l’X fragile, la forme la plus fréquente de retard mental héréditaire, entraîne l'assemblage spatial des nucléoporines, des protéines assurant la fonction et la survie de la cellule. Une avancée qui pourrait à terme ouvrir de nouvelles perspectives thérapeutiques.
 Arantxa Agote Aran, doctorante,
Arantxa Agote Aran, doctorante, et Izabela Sumara travaillent sur
le syndrome de l'X fragile. Photo MR
Izabela Sumara s’intéresse depuis 20 ans à la division cellulaire. Durant ses recherches, la biologiste cellulaire découvre une protéine dont la fonction exacte n’est alors pas connue. « Nous avons été étonnés de constater qu’elle interagissait avec la famille de protéines liées au X fragile à savoir FXR1, FXR2 et FMRP, qui ont un rôle dans la pathologie du même nom. »
Epaulée par Jean-Louis Mandel, ancien chercheur de l’IGBMC qui a découvert le rôle de cette famille de protéines dans le syndrome de l’X fragile, elle remarque que ces protéines sont localisées dans les cellules au niveau de l’enveloppe nucléaire, une structure à double membrane entourant le noyau de chaque cellule.
Les protéines liées au X fragile préviennent l’agrégation aberrante des nucléoporines
Au niveau de cette membrane se trouvent également les nucléoporines, des protéines appartenant aux nucléopores qui assurent le transport et l'échange de nombreuses molécules essentielles entre le noyau et le cytoplasme. Reste à déterminer la relation qui unit les protéines de l’X fragile et ces nucléopores.
Résultat : En enlevant les protéines de l’X fragile des cellules, la chercheuse et son équipe remarquent que les nucléoporines s'assemblent de manière aberrante dans le cytoplasme en structures semblables à des agrégats. La fonction des nucléopores est également affectée. « Il y avait des défauts au niveau du transport du noyau vers le cytoplasme. »
Izabela Sumara se demande alors si ces caractéristiques sont les mêmes dans les modèles du syndrome de l’X fragile caractérisé par l’absence de la protéine FMRP. « Il est intéressant de noter que les modèles de ce syndrome montrent les mêmes agrégats aberrants de nucléoporines cytoplasmiques. Cela suggère que ces défauts peuvent contribuer à cette pathologie. »
Vers de nouvelles stratégies thérapeutiques
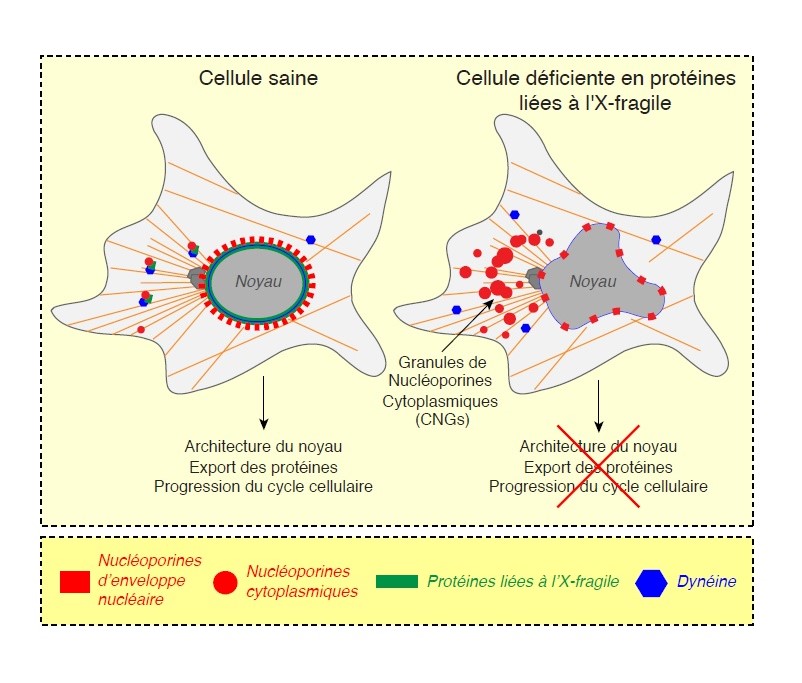 En l'absence des protéines de l’X fragile,
En l'absence des protéines de l’X fragile, les nucléoporines s'assemblent de manière
aberrante dans le cytoplasme. Crédit DR
Cette famille de protéines était étudiée depuis longtemps. « Mais c’est la première fois que nous déterminons leur rôle dans la localisation des nucléoporines. Nous pensons que l’inhibition de l’export des protéines joue un rôle déterminant dans le développement, en particulier sur les neurones. »
Prochaine étape : trouver des facteurs pour contrecarrer l’agrégation des nucléoporines dans le cytoplasme. A long terme, cela pourrait permettre de développer de nouvelles stratégies thérapeutiques. « Ce qui est intéressant aussi c’est que l’agrégation des nucléoporines est caractéristique d’autres maladies comme Alzheimer ou Huntington. Cette découverte est une nouvelle pièce au puzzle qui pourrait contribuer à la recherche contre ces maladies. »
Marion Riegert
- Retrouvez la publication du 24 juillet 2020 dans The Embo journal.
Le syndrome de l’X fragile
Get more information
Le syndrome de l’X fragile est une maladie neurologique qui est la première cause de retard mentale héréditaire. Les personnes atteintes sont également plus susceptibles de développer de l’autisme, une malformation faciale ou encore de l’infertilité. Il n'existe pas de remède à ce jour. Le syndrome doit son nom au gène muté dans la pathologie. Localisé sur le chromosome X, ce dernier va former une structure particulière comme s’il y avait une cassure. Cette forme chromosomique particulière a longtemps été un marqueur de pronostic de la maladie.